Marcado CE de productos sanitarios
El nuevo reglamento de productos sanitarios
Bienvenido a la columna vertebral de la conformidad normativa en dispositivos médicos: el Reglamento de Productos Sanitarios (MDR). En Alenta Consulting, desentrañamos este complejo marco regulatorio para que comprendas su esencia. El MDR, establece estándares rigurosos para garantizar la seguridad y el rendimiento de los dispositivos médicos en la Unión Europea. Desde su alcance geográfico hasta los requisitos esenciales para la colocación en el mercado, nuestra guía detallada te proporciona una visión completa. Te invitamos a explorar este universo regulatorio con nosotros, donde la claridad y la certeza te guiarán hacia el cumplimiento efectivo.
Diagrama de tiempos del MDR
En este viaje hacia el cumplimiento normativo, es crucial comprender los tiempos de aplicación del MDR. No es solo un marco estático; es un proceso en constante evolución. En Alenta Consulting, te ofrecemos una sección dedicada a analizar los plazos críticos. Desde la fecha de aplicación hasta los períodos de transición, te guiamos para que planifiques estratégicamente y evites sorpresas regulatorias. Mantén tu camino hacia el cumplimiento claro y bien definido con nuestra experiencia a tu disposición.
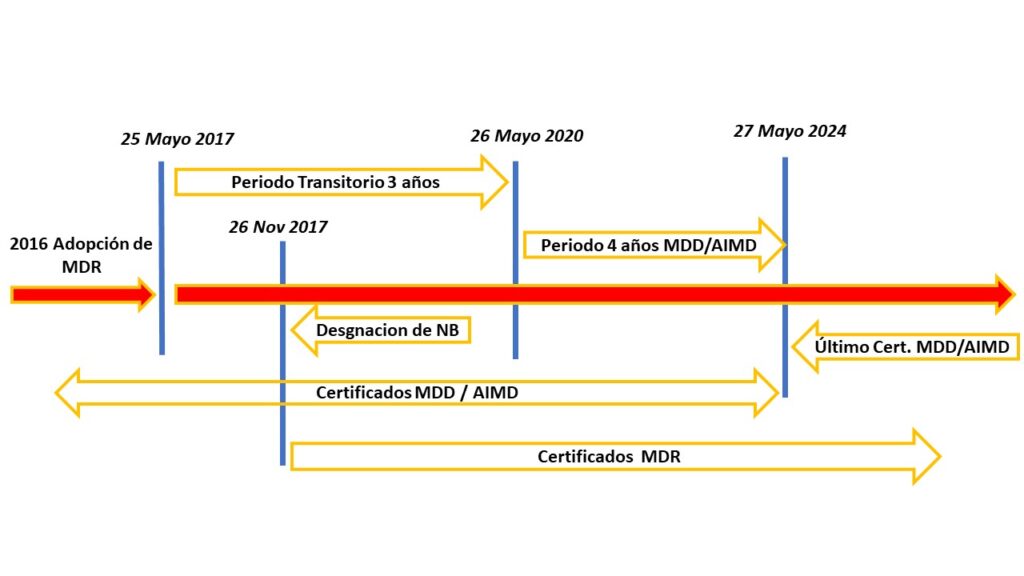
Aspectos importantes del MDR
Requisitos Generales de Seguridad y Funcionamiento (GSPR)
En el corazón de la conformidad normativa se encuentran los Requisitos Generales de de Seguridad y Funcionamiento (GSPR). Estos criterios establecen los estándares de seguridad y rendimiento que tu producto debe cumplir. En Alenta Consulting, te sumergimos en los detalles, proporcionándote una visión detallada de los GSPR y su impacto en tu producto. Desde la evaluación de riesgos hasta los requisitos de diseño, te acompañamos para garantizar que tu producto no solo cumpla, sino supere las expectativas regulatorias.
El MDR en su Anexo I establece los Requisitos Generales de Seguridad y Funcionamiento o GSPR (General Safety Performance Requirements) y los divide en:
- Requisitos generales (Capítulo I)
- Requisitos relativos al diseño y la fabricación (Capítulo II)
- Requisitos relativos a la información proporcionada con el producto (Capítulo III)
Los fabricantes están obligados a cumplirlos para demostrar la conformidad con el MDR y deben disponer de una documentación especifica que justifique dicho cumplimiento.
Puede encontrar más información en General Safety Performance Requirements
Clasificación de Productos Sanitarios (MD)
Abordamos la clasificación de productos sanitarios, un proceso clave en el cumplimiento del MDR. Desde dispositivos de bajo riesgo hasta aquellos con impacto significativo, analizamos cómo la finalidad prevista afecta la categorización. Comprender estos elementos fundamentales es esencial para desarrollar una estrategia efectiva de cumplimiento normativo.
Antes de clasificar un producto sanitario el fabricante debe tener claro cual es la finalidad prevista.
La definición de finalidad prevista está establecida en el artículo 2 apartado 12 del MDR:
12) «finalidad prevista»: el uso al que se destina un producto según los datos facilitados por el fabricante en el etiquetado, en las instrucciones de uso o en el material o las declaraciones de promoción o venta, y según lo indicado por el fabricante en la evaluación clínica;
Una vez definida será fácil saber si el producto cumple con la definición de producto sanitario tal y como recoge el artículo 2, apartado 1 del MDR.
1) «producto sanitario»: todo instrumento, dispositivo, equipo, programa informático, implante, reactivo, material u otro artículo destinado por el fabricante a ser utilizado en personas, por separado o en combinación, con alguna de las siguientes finalidades médicas específicas:
-diagnóstico, prevención, seguimiento, predicción, pronóstico, tratamiento o alivio de una enfermedad,
-diagnóstico, seguimiento, tratamiento, alivio o compensación de una lesión o de una discapacidad,
-investigación, sustitución o modificación de la anatomía o de un proceso o estado fisiológico o patológico,
-obtención de información mediante el examen in vitro de muestras procedentes del cuerpo humano, incluyendo donaciones de órganos, sangre y tejidos, y que no ejerce su acción principal prevista en el interior o en la superficie del cuerpo humano por mecanismos farmacológicos, inmunológicos ni metabólicos, pero a cuya función puedan contribuir tales mecanismos.
Los siguientes productos también se considerarán productos sanitarios:
— los productos de control o apoyo a la concepción,
— los productos destinados específicamente a la limpieza, desinfección o esterilización de los productos que se contemplan en el artículo 1, apartado 4, y en el párrafo primero del presente punto;
Si el producto en cuestión fuera MD el siguiente paso sería obtener su clasificación tal y como recoge el MDR en su artículo 51.
El Anexo IX establece las reglas que hacen que los productos sanitarios caigan en una u otra clasificación. Las clases son:
- Clase I / Clase I con función de medición / Clase I estéril
- Class IIa
- Class IIb
- Class III
Puede encontrar más información en Finalidad prevista
Puede encontrar más información en Reglas y clasificación de productos sanitarios
Puede encontrar más información en Clasificación del SW
Documentación Técnica
Entramos en el mundo de la documentación técnica, un componente crítico en el proceso de conformidad. Desde el expediente técnico hasta los informes de evaluación clínica, exploramos la documentación esencial requerida por el MDR. Simplificamos este proceso complejo, brindándote orientación paso a paso.
El MDR en su art. 10 establece que el fabricante deberá mantener la documentación técnica durante al menos 10 años después de que la ultima unidad de producto sanitario haya sido comercializada y avalada por la declaración de conformidad que le correspondiera. La documentación técnica debe estar presentada de una forma clara, organizada, fácilmente localizable y sin ambigüedades y deberá incluir:
- Descripción del dispositivo, especificación incluyendo las variantes y los accesorios
- Etiquetado y embalaje del producto
- Instrucciones de uso
- Información completa de diseño y fabricación
- La lista de los requerimientos generales de seguridad y funcionamiento (GSPR) que sean de aplicación, así como las normas de aplicación, los comentarios y los documentos que soportan la aplicabilidad o no del requerimiento.
- Análisis de riesgo y conclusiones sobre la relación beneficio/riesgo
- Verificación y validación del producto
- Documentación del proceso de vigilancia post comercialización
Puede encontrar más información en Documentación técnica
Sistema de identificación única y etiquetado (UDI)
Enfrentamos el desafío de la Identificación Única de Dispositivos (UDI). Explicamos en detalle cómo implementar eficazmente el sistema UDI, destacando su importancia en la trazabilidad de productos. Con nuestra experiencia, garantizamos que tu producto cumpla con los estándares de identificación exigidos por el MDR. El MDR en su art. 27 y 28 establecen la creación de una base de datos del sistema de identificación única (UDI), es decir, un sistema de nomenclaturas que permite la identificación y la trazabilidad del producto sanitario a lo largo de todo su ciclo de vida.
El fabricante está obligado antes de la comercialización, a asignar un UDI y lo usará para tanto las consultas como para las actualizaciones en la base de datos EUDAMED. Este UDI deberá ser colocado en la etiqueta del producto y ser registrado.
UDI está relacionado con el Sistema de Nomenclatura Europea de Productos Sanitarios o EMDN por sus siglas en inglés de European Medical Device Nomenclature.
El código EMDN está formado por 7 niveles de información, aunque existen 3 niveles principales: categoría, grupo y tipos. El nivel «tipos» se puede descomponer en los niveles 4,5,6 y 7 dependiendo de si son necesarios para la codificación del tipo de producto.
Puede encontrar más información en UDI
Puede encontrar más información en UDI en SaMD
Puede encontrar más información en EMDN
Base de datos EUDAMED
Exploramos la Base de Datos EUDAMED, la piedra angular de la transparencia y la gestión de datos bajo el MDR. Te guiaremos a través de su funcionalidad y cómo interactuar eficientemente con esta plataforma centralizada. Aprovecha al máximo esta herramienta para optimizar la gestión de datos y cumplir con los requisitos regulatorios.
Los artículos 27 y 28 establecen la creación de una BBDD denominada EUDAMED sobre los productos sanitarios.
EUDAMED estará compuesta por seis módulos relacionados con:
- agentes económicos: fabricantes de la UE y de fuera de la UE, representantes autorizados, , un distribuidor o entidades que realizan un reprocesamiento de un producto
- UDI y registro de productos
- Organismos notificados y certificados
- Investigaciones clínicas y estudios de seguridad
- Vigilancia
- Seguimiento post-comercialización
Puede encontrar más información en EUDAMED
Nuevos roles
MDR en su art. 15 establece la necesidad de una persona responsable de la normativa con una competencia determinada, que vendrá dada o bien por experiencia o bien por titulación con menos experiencia, pero que en cualquier caso será responsable de:
- Antes de liberar un producto verificar la conformidad con el sistema de calidad implantado en su fabricación
- Actualizar las declaraciones de conformidad de los productos, así como la documentación técnica.
- Cumplir con las obligaciones de seguimiento post comercialización
- Notificar los incidentes de seguridad dentro del procedimiento de vigilancia
- Cuando se trate de productos de investigación, hacer las declaraciones pertinentes establecidas en el Anexo XV, Capitulo II sección 4.1
Esta persona no tiene por qué ser empleado de la organización, pudiendo ser un consultor o una persona subcontratada a tal fin.
Vigilancia en el mercado o Post Market Surveillance
Sumérgete en las aguas de la vigilancia poscomercialización. Analizamos la importancia de la vigilancia en el mercado y cómo cumplir con este aspecto crítico del MDR. Desde la recopilación de datos hasta la notificación de incidentes, te proporcionamos las herramientas necesarias para garantizar la seguridad continua de tus productos. Para cada producto el fabricante debe planificar, establecer y mantener un sistema documental sobre el seguimiento post comercialización PMS (del inglés Post Market Surveillance) que además formará parte del SGC de la empresa.
El principal objetivo que se pretende es que el fabricante sea capaz de recabar, registrar y analizar toda la información del producto durante todo su ciclo de vida, para poder obtener conclusiones sobre su desempeño.
Un sistema de PMS deberá constar de:
- Planes:
- PMS Plan que nos servirá para establecer las responsabilidades, objetivos, fuentes de los datos, qué métodos, indicadores y niveles de umbral usaremos. Igualmente con el estableceremos cual es la gestión que haremos de incidentes no serios y efectos secundarios indeseables, plazos y frecuencia de actualización, el modo en el que recogeremos los datos y las referencias a documentación tanto interna como externa.
- PMCF Plan. PMCF viene del inglés Post Market Clinical Follow-up. El PMCF es el seguimiento clínico que haremos del producto después de su comercialización y genera inputs relevantes para la Evaluación clínica.
- Informes:
- PMSR o PSUR. Son el Post Market Surveillance Report o el Periodic Safesty Update Report que recogen las conclusiones que hemos obtenido sobre el PMS según lo planificados
- PMCF Report. Son las conclusiones que hemos recogido desde un punto de vista clínico acerca del producto.
- SSCP. El Summary of Safety and Clinical performance, que resumirá lo que diga el PMSR o PSUR y el PMCF report.
Puede encontrar más información en Vigilancia en el mercado
Evaluación clínica
Afronta la evaluación clínica con confianza. En esta sección, desglosamos los requisitos y mejores prácticas para garantizar una evaluación clínica sólida y conforme al MDR. Desde la planificación hasta la presentación, nuestra experiencia en evaluación clínica elevará la calidad y la confianza en tus productos. Es el proceso obligatorio, sistemático y planificado para generar, recoger, analizar y evaluar de forma continua los datos clínicos relativos a un producto para verificar su seguridad y funcionamiento, incluidos los beneficios clínicos, cuando se utilice conforme a la finalidad prevista por el fabricante.
Tiene estos objetivos:
- Demostrar el beneficio clínico del producto.
- Asegurar que no habrá efectos no esperados o efectos secundarios no aceptables.
Además es absolutamente necesaria para realizar el Análisis de Riesgos, ya que solamente la evaluación clínica justifica las aceptaciones / no aceptaciones del nivel de riesgo obtenido en dicho análisis.
La evaluación clínica es una parte esencial de los GSPR del Anexo I del MDR. En su artículo 5, apartado 3 se menciona explícitamente que:
- La demostración de la conformidad con los requisitos generales de seguridad y funcionamiento deberá incluir una evaluación clínica con arreglo al artículo 61.
De acuerdo con el artículo 61 del MDR una evaluación clínica puede ser definida como un proceso metodológico basado en lo siguiente:
- una evaluación crítica de literatura científica relevante
- una evaluación crítica de los resultados de las investigaciones clínicas disponibles
- la consideración de un tratamiento alternativo disponible en ese momento para el mismo fin si las hubiera.
Se realizará un informe de la Evaluación Clínica o lo que se conoce como Clinical Evaluation Report (CER). Es informe formará parte de la documentación técnica del producto.
Puede encontrar más información en Evaluación clínica
Puede encontrar más información en Evaluación clínica de SW como producto sanitario.
