El fabricante preparará una documentación técnica según Anexo II para su producto sanitario que usará para dar cumplimiento con los GSPR del Anexo I.
El fabricante preparará una documentación técnica sobre su productos que será: clara, organizada, fácilmente localizable y sin ambigüedades. Cada fabricante lo organiza como desea aunque aquí se presenta una manera de hacerlo ordenada de la misma manera que se establece en el MDR y mas abajo pasamos a comentar cada uno de los puntos:
0. Guía
Será buena práctica incluir una pequeña descripción a modo de indice en la que se explique brevemente cada uno de los puntos así como la lista de documentos que se relacionan con cada uno de los puntos anteriores. Igualmente indicaremos la versión del documento que está en vigor.
1. Descripción del producto y especificaciones.
Describiremos el producto de forma general incluyendo su finalidad prevista así como los usuarios previstos para el producto. Daremos referencias de datos como: categoría de producto, modelo o código de producto, nombre comercial o marca comercial. Igualmente indicaremos el UDI-DI que tiene nuestro producto y que el fabricante ha asignado previamente mediante el uso de la nomenclatura EMDN. Aspectos interesantes que tenemos que mencionar son el grupo de pacientes destino para quien se realizará el diagnostico, tratamiento o monitorización. En cada uno de los apartados o documentos se puede hacer referencia a otros documentos como por ejemplo: Instructions for Use (IFU), Clinical Evaluation Report, etc).
La Declaración de Conformidad o DoC del inglés Declaration of Conformity, será incluida en este apartado siendo este documento merecedor de un artículo especifico por la importancia de su significado.
Incluiremos asi mismo, cual es la clasificación de MD de acuerdo con las reglas del MDR y daremos las oportunas justificaciones de la clasificación realizada. Haremos referencia igualmente a los accesorios del MD o sin serlo, están específicamente destinados a usar en combinación con el.
Una lista de variantes o posibles configuraciones que puede tener el MD y que están destinadas a ser puestas en el mercado. Igualmente será interesante hacer una lista de todas las materias primas que han sido usadas para la fabricación, así como si tienen contacto con el cuerpo o no.
Haremos referencia a aquellos MD de generaciones anteriores que ya no se pongan en el mercado pero que sean los predecesores del mismo y también aquellos productos que sean similares de otros fabricantes.
2. Información suministrada por el fabricante
Las etiquetas del MD o su envase o embalaje así como las condiciones de transporte o almacenaje. Habrá que crear un plan de etiquetado de los productos y tener en cuenta los símbolos estandarizados de la norma ISO 15223:2021.
Las IFUs (instruction for use) en una lenguas de los estados miembros así como en la lengua del pais en la que se vaya a vender el MD. Incluirá información relativa entre otros al grupo de pacientes destino, a las indicaciones y contraindicaciones. Habrá una referencia al SSCP (Summary of Safety Clinical Performance) y a los beneficios clínicos, a la formación que debe tener el personal que use el MD. Si el MD tuviera como finalidad prevista su uso en operaciones quirúrgicas, habrá que especificar cuales son esas técnicas. Pueden estar incluidas dentro del manual de usuario o IFU. Es importante el material promocional que se use y como está redactado para no causar malentendidos.
3. Diseño y fabricación
El lugar donde se realiza el diseño y/o la fabricación son importantes en este proceso principalmente porque pueden llegar a ser auditados por el Organismo Notificado. Las condiciones ambientales/físicas/biológicas/higiénicas serán en algunos casos decisivas para la fabricación o proceso de un MD y el Organismo Notificado querrá verificar el cumplimiento con el MDR.
El diseño es una parte importante del proceso de desarrollo de un MD. Así, deberemos planificar cada una de las etapas del desarrollo y mantener esos planes actualizados. Indicaremos qué metodologías hemos usados, qué herramientas de planificación, las especificaciones de partida y de donde las hemos obtenido, como integramos nuestro producto con el entorno si fuera el caso, como verificamos que el producto cumple con la normativa de aplicación, como analizamos el riesgo existente en nuestro producto y como mitigamos dichos riesgos. Está claro que si el producto incluye SW y HW ambas partes deberán estar muy bien documentadas.
4. General Safety and Performance Requirements (GSPR)
Normalmente para dar conformidad con los GSPR usaremos normas armonizadas, que son las que dan presunción de conformidad pero es el fabricante el único que decide si usarlas o no.
La recomendación es usarlas ya que de lo contrario, habrá que justificar las que usemos. Como práctica habitual los fabricantes las usan porque de esta forma los Organismos Notificados pueden evaluar la conformidad de una forma más rápida y cómoda. A continuación aparece una imagen obtenida del último listado de normas armonizadas para el MDR (modificación de la Decisión (EU) 2021/1182 de 17/05/2022). Se puede observar que para la implantación de un SGC para MD tendremos que usar la norma EN ISO 13485:2016+AC:2018+A11:2021 y para realizar el análisis de riesgos usaríamos la norma EN ISO 14971:2019+A11:2021.

Un ejemplo de como deberíamos completar la lista de GSPR puede ser este:

5.Gestión y Análisis de riesgos
La gestión del riesgo es algo que debe formar parte del ciclo de vida completo del producto y ademas debe actualizarse con cada nueva contemplar desde las fases iniciales del desarrollo de un MD. Cuando hablamos de riesgo a lo que nos estamos refiriendo es a la probabilidad de que un evento se produzca multiplicado por el impacto que tiene ese evento. Por lo que es muy importante que el fabricante tenga definida cual es su política de riesgos así como los criterios para aceptar un posible riesgo.
Para definir bien esta parte de la documentación que tenemos que preparar, hay que empezar con un PHA (Preliminary Hazard Analysis), que es un estudio preliminar de los peligros que pueden existir basándonos en sistemas que ya existan o en el nuestro propio, y que identificarán los principales peligros así como las probabilidades de que ocurran.
A continuación podemos preparar el Risk Management Plan en el que definiremos aspectos como qué metodología usaremos, qué inputs y qué origen tienen o qué criterios de aceptación usaremos por ejemplo.
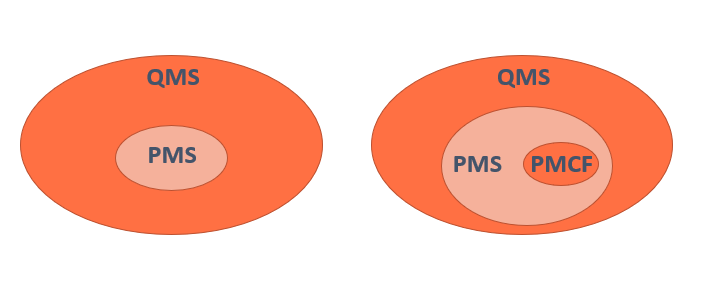
La gestión del riesgo está íntimamente relacionada con la evaluación clínica, de la que obtiene información para añadir nuevos riesgos o modificarlas si fuera el caso.
Para realizar nuestra gestión y análisis de riesgos usaremos la norma EN ISO 14971. Del mismo modo debemos explicar en un informe los resultados y argumentar las decisiones acerca de la relación beneficio/riesgo.
6. Verificación y validación del producto
El MDR establece en el Anexo II Apartado 6, que se deberán documentar todos los ensayos y test de validación que se realice para demostrar la conformidad del producto con los GSPR.
Esta documentación sobre validación y verificación contendrá aspectos como:
- estudios de biocompatibilidad
- características químicas o físicas
- requisitos de seguridad eléctrica o compatibilidad electromagnética.
- condiciones de almacenaje o transporte
- aspectos de usabilidad o reusabilidad
- etiquetado y simbología usada adecuada
- como aseguramos la exactitud en las medidas.
Usando datos clínicos, tendremos que elaborar el Informe de Evaluación Clínica o CER del inglés Clinical Evaluation Report. Para mas información puede visitar este artículo.